Working with cluster queuing systems
Overview
Teaching: 0 min
Exercises: 0 minQuestions
Key question (FIXME)
Objectives
First learning objective. (FIXME)
Supercomputers are actually a collection of many smaller computers loosely tied together, this is why they are often referred to as clusters. When we ssh in to the cluster, we are actually logging in to a specific computer in the cluster called a login node. These computers are designed to communicate from the cluster to the outside world, but they are not designed for doing intensive computation. There are separate computers called compute nodes which are used for running jobs.
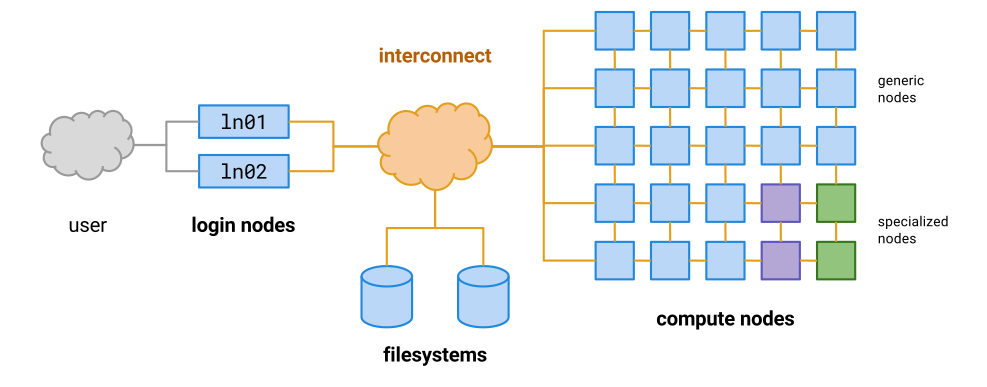
Instead of doing our work directly on the login node, as we have been doing in the previous lessons, we should instead write scripts that contain the commands we want to run, and then use the cluster’s queueing system to allocate those commands to a free computer within the cluster (a compute node).
Let’s test this out by writing a very simple script.
nano test-qsub.sh
Add the following commands to your new script:
hostname
whoami
pwd
This script should print the following information:
- The name of the computer it is running on
- The name of the user that is running it
- The current working directory
Let’s test it out on the login node by asking bash to run it for us:
bash test-qsub.sh
deva
username
/home/username/chip-tutorial/
Hopefully the output should be what you expected. Let’s now submit the script to the cluster and ask
it to allocate these commands to a free compute node. We can do this using qsub.
qsub test-qsub.sh
Your job 3490362 ("test-qsub.sh") has been submitted
While our job is running, we can check its status using qstat:
qstat
The output is a long list of every job that’s running on the system, which can be a little difficult
to navigate. To make things easier, we can use grep to search specifically for our own jobs.
qstat | grep username
Once our job has finished, we need to find the output. Let’s check our current directory:
ls -l
drwxr-xr-x 2 user group 24 May 6 15:33 aligned_reads
drwxr-xr-x 2 user group 2 May 5 15:25 annotation
drwxr-xr-x 2 user group 8 May 11 11:49 bigwigs
drwxr-xr-x 2 user group 6 Apr 29 13:53 bowtie2-index-mm10-chr11
drwxr-xr-x 2 user group 19 May 4 15:56 fastqs
-rw-r--r-- 1 user group 20 May 12 09:37 test-qsub.sh
There are no new files there. Instead, the output has been saved in our home directory in a file that starts with the same name as our script:
ls -l ~/test-qsub.sh*
-rw-r--r-- 1 user group 0 May 12 09:37 /home/group/user/test-qsub.sh.e3490361
-rw-r--r-- 1 user group 50 May 12 09:37 /home/group/user/test-qsub.sh.o3490361
Let’s open the output file (.o) and see the contents.
cat ~/test-qsub.sh.o3490361
cbrgbigmem01p
user
/home/user
The first line shows that these commands were not run on the login node, although they were run using our user account. They also weren’t run in the directory of the script, but rather in our home directory. We almost always want our jobs to run in the directory of the script file, but we need a way to explicitly tell the cluster this is what we want. We can add it as an option to our script file:
nano test-qsub.sh
#$ -cwd
hostname
whoami
pwd
Now let’s submit the job again:
qsub test-qsub.sh
Your job 3490362 ("test-qsub.sh") has been submitted
qstat | grep username
ls -l
drwxr-xr-x 2 user group 24 May 6 15:33 aligned_reads
drwxr-xr-x 2 user group 2 May 5 15:25 annotation
drwxr-xr-x 2 user group 8 May 11 11:49 bigwigs
drwxr-xr-x 2 user group 6 Apr 29 13:53 bowtie2-index-mm10-chr11
drwxr-xr-x 2 user group 19 May 4 15:56 fastqs
-rw-r--r-- 1 user group 29 May 12 10:14 test-qsub.sh
-rw-r--r-- 1 user group 0 May 12 10:14 test-qsub.sh.e3490364
-rw-r--r-- 1 user group 57 May 12 10:14 test-qsub.sh.o3490364
This time the output has been saved in our current directory. Let’s check the output file:
cat test_qsub.sh.o3490364
cbrgwn006p
user
/home/user/chip-tutorial
The script ran on a different compute node this time (maybe it didn’t for you), and it ran in the folder where the script is, which is much more convenient.
Other qsub script parameters
There are many other parameters for scripts that you are going to submit via qsub that you might find useful. These should all be on new lines that start with #$
-o /different/output/file: Save script output to this file-e /different/error/file: Save script error messages to this file-N job_nameGive your job a different name to find it easier in qsub-m eaSend an email when the job finishes-l h_vmem=20GAsk the queue to allocate more memory for the job (this may be necessary for mapping jobs)So for example a typical script might start like this:
#$ -cwd #$ -o job_output.txt #$ -e job_errors.txt #$ -N job_name #$ -m ea
Making bigwigs using the cluster queue
In the last session we made bigwig files that covered around 100kb of chromosome 11, because making them for the whole chromosome would have taken too long. Now we can write a script to make the full bigwigs and submit it to the cluster using qsub.
Make a script called
make_bigwigs.shand edit it so that it makes all eight bigwigs.HINT: Think about why we need two for loops for this, rather than just one?
HINT: This script uses the
basenamecommand, which we haven’t met before. Try it out in the terminal to figure out what it does.#$ -cwd #$ -m ea module load deeptools/3.0.1 for bam in aligned_reads/G1E_ATAC*.sorted.bam do bamCoverage ____ -o bigwigs/$(basename $bam | cut -d "." -f 1,2).bw -r chr11 done for bam in aligned_reads/G1E_ChIP*.sorted.bam do bamCoverage ____ -o bigwigs/$(basename $bam | cut -d "." -f 1,2).bw -r chr11 doneSolution
#$ -cwd #$ -m ea module load deeptools/3.0.1 for bam in aligned_reads/G1E_ATAC*.sorted.bam do bamCoverage -b $bam --binSize 1 \ --blackListFileName annotation/mm10-blacklist.ENCSR636HFF.v2.bed \ --normalizeUsing RPKM --extendReads -o bigwigs/$(basename $bam | cut -d "." -f 1,2).bw -r chr11 done for bam in aligned_reads/G1E_ChIP*.sorted.bam do bamCoverage -b $bam --binSize 1 \ --blackListFileName annotation/mm10-blacklist.ENCSR636HFF.v2.bed \ --normalizeUsing RPKM --extendReads 150 -o bigwigs/$(basename $bam | cut -d "." -f 1,2).bw -r chr11 done
If we want to run a few short commands using the cluster, we can also use qlogin instead of qsub
Key Points
First key point. Brief Answer to questions. (FIXME)